Emulating explicit chemistry of organic aerosols using machine learning
Natural and anthropogenic sources emit a large number of volatile organic compounds (VOCs) which can get oxidized in the atmosphere leading to hundreds of organic compounds that contribute to the formation of secondary organic aerosols. Accurate predictions of the complex chemistry and distribution of these gaseous and particle phase species is key for air quality and climate studies due to aerosol effects on both solar radiation and cloud formation. Explicit models such as the Generator for Explicit Chemistry and Kinetics of Organics in the Atmosphere (GECKO-A) are needed to accurately represent the atmospheric chemical processing of organic species, however these models are computationally expensive and thus cannot be used in chemistry-climate simulations.
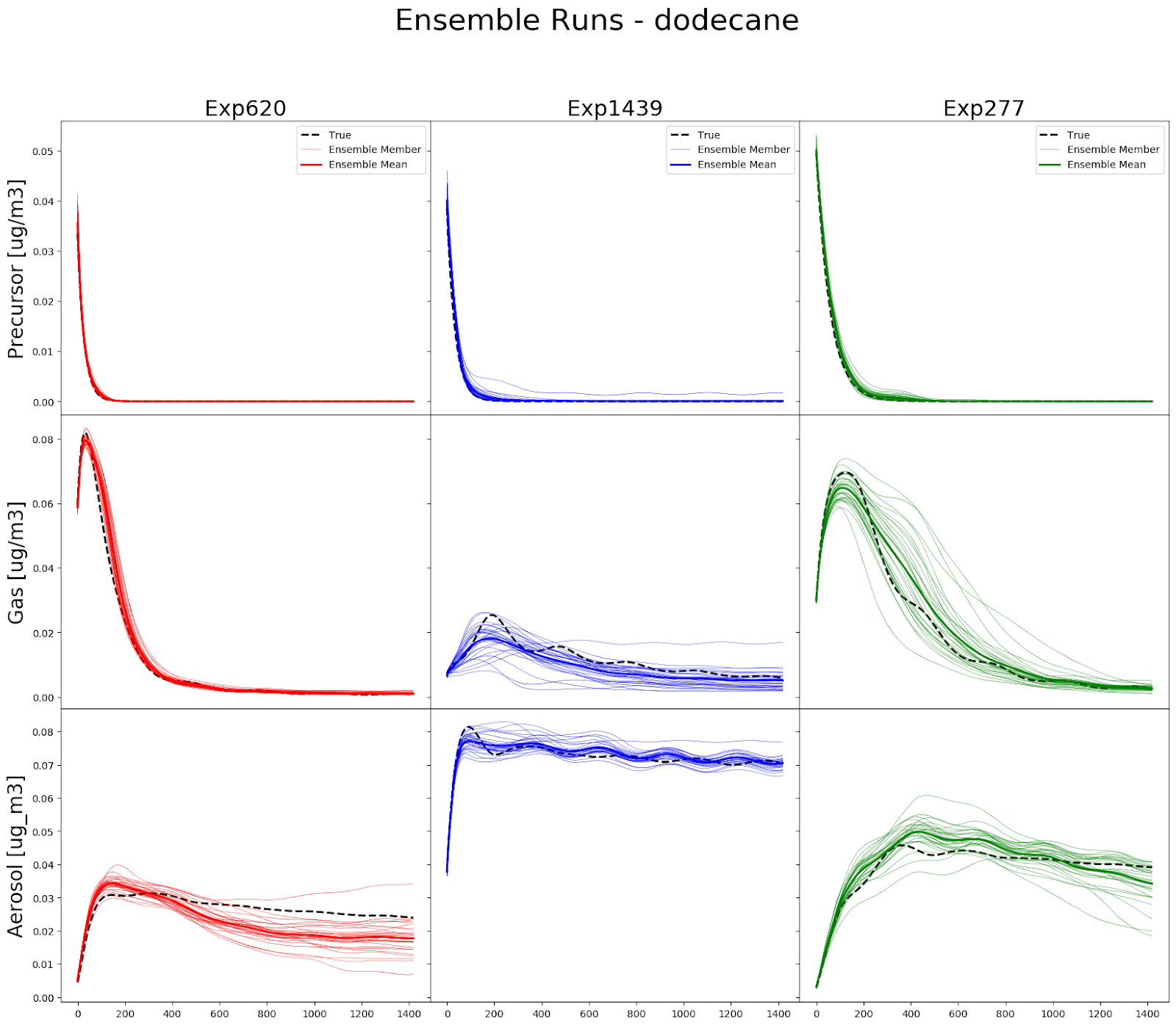
Recent applications of atmospheric chemistry emulation for gas-phase chemistry have shown its ability to significantly reduce the computational costs (Keller et al., 2019). We have investigated whether these techniques can be applied to organic aerosol chemistry to build robust parameterizations for use in 3D models. For that purpose, we have generated an open-benchmarking dataset using the GECKO-A explicit model. It includes 2,000-5,000 five-day simulations for several organic aerosol precursors. We have used this dataset to provide some initial benchmarks using a variety of machine learning methodologies on a forward walking box model (Figure 1). These include both recurrent neural networks, and single time-step models with recurrent training, which have been shown to reduce numerical instability. We have demonstrated that the machine learning algorithm can be built for 3 individual precursor species coming from very different emission sources – toluene from anthropogenic emissions, C12 n-alkane from traffic/urban emissions and terpenes from forest emissions. This was done for fixed idealized conditions (temperature, radiation, chemical composition). The next steps of this project will focus on expanding this framework to incorporate mixtures of urban precursors under ambient conditions where temperature and ambient chemical species vary during the day.
References
Keller, C. A. and Evans, M. J.: Application of random forest regression to the calculation of gas-phase chemistry within the GEOS-Chem chemistry model v10, Geosci. Model Dev., 12, 1209–1225, https://doi.org/10.5194/gmd-12-1209-2019, 2019.