Secondary Organic Aerosols (SOA)
Secondary Organic Aerosol (SOA) formation in aqueous particles
Recent research suggests that in-cloud and in-particle chemistry could contribute substantially to the formation of SOA. Glyoxal is one of the precursors proposed to be important. In Knote et al. (2013) we included the state of knowledge on SOA formation from glyoxal into WRF-chem and conducted simulations over California as well as the continental United States for summer 2010. We found that over California the South California Air Basin is the hotspot for SOA formation from glyoxal, due to the high availability of precursors, aerosols and humidity. The eastern slopes of the Central Valley with their high isoprene loadings did not show substantially elevated levels of glyoxal SOA, despite the fact that isoprene is a major precursor for glyoxal. This is because of rather dry conditions that introduce kinetic limitations in the rate of glyoxal-SOA formation (Kampf et al., 2013). We found that formation chemistry in the bulk liquid phase of aerosols leads on average to a 1% contribution to total SOA mass, while a simple parameterization under the assumption of a surface process suggests that up to 15% of total SOA mass is actually from glyoxal in the South California Air Basin. Reversible partitioning of glyoxal into the aerosols (e.g. through oligomerization) has been found to be negligible in its mass contribution. The reason for the lower SOA mass yields when looking at volume chemistry does not seem to be caused by slow in-particle chemistry, but rather limited by the efficiency to transfer glyoxal from the gas-phase into the aerosols. The Eastern US shows high potential for SOA formation from glyoxal due to very high amounts of precursors, aerosols and humidity (see Figure 1).
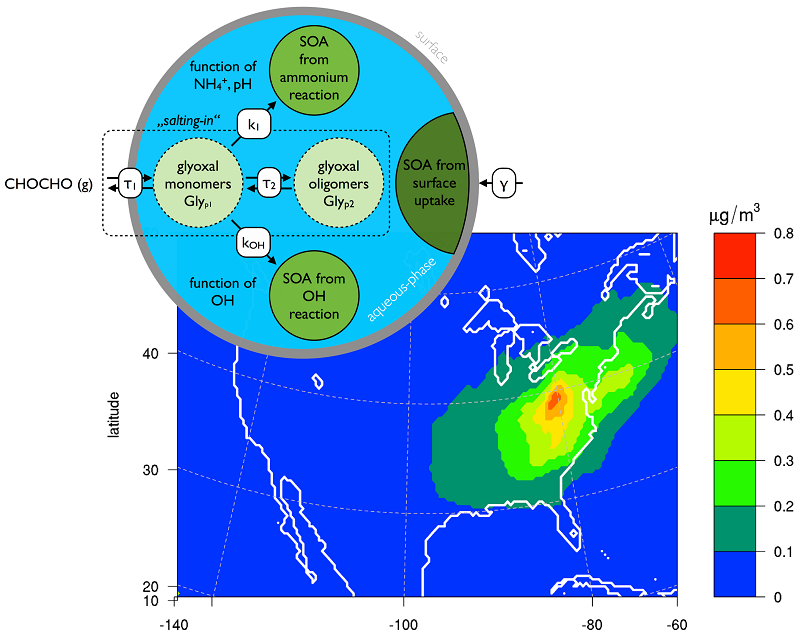
Figure 1. Upper left shows a schematic of glyoxal (CHOCHO) transformations into and within the liquid phase of aerosols. Lower panel shows the mass concentration of secondary organic aerosol produced from glyoxal chemistry within the aerosol liquid phase.
Effects of dry and wet deposition of organic vapors on Secondary Organic Aerosol (SOA)
Dry and wet deposition are the main removal pathways for SOA. Wet scavenging is typically found to be the major direct loss (90%) of SOA in chemistry climate models (Tsigaridis et al. 2013), while dry deposition of OA represents a much smaller sink (<10%) due to low dry deposition velocities of aerosols. SOA loss can also occur indirectly by wet and dry removal of semi-volatile gas-phase oxidation intermediates (SVOCs) which can suppress the amount of condensable material available for SOA formation or lead to evaporation of SOA to satisfy the thermodynamic equilibrium. The effect of this indirect removal on SOA surface concentrations is highly uncertain as 3D models typically consider gas-phase intermediates as insoluble, or use a specified Henry’s law solubility coefficient. New analysis by Hodzic et al. (2014) based on detailed calculations using explicit oxidation chemistry suggests however that Henry’s law solubility coefficients can vary significantly as a function of SVOC volatility, suggesting the need for reassessment of effective wet and dry removal lifetimes of SOA. Inclusion of this process in the WRF-Chem model shows that deposition of partly-oxygenated organic gases is a major loss of organic particles, as these evaporate to re-equilibrate with the changing gas concentrations (Knote et al., 2014). Averaged over the U.S., this additional removal process leads to large reductions in predicted surface OA concentrations (see Figure 2).
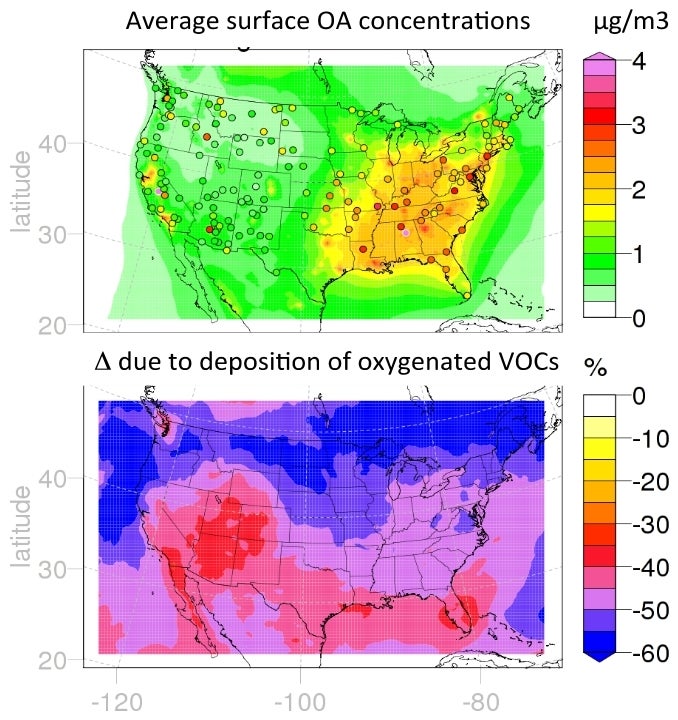
Figure 2. Upper panel is the summer average WRF-Chem model surface organic aerosol (OA) concentrations at the surface. The color dots are OA measurements from the IMPROVE network. Lower panel is the change in OA concentrations due to the inclusion of dry and wet removal of condensable organic vapors calculated from the WRF-Chem model.