Modeling of SOA formation using an explicit gas-phase chemical mechanism
The mechanisms by which secondary organic aerosols (SOA) form in the atmosphere are a topic of much current research. Parameterizations used in air quality and climate have difficulty reproducing observed quantities of ambient aerosol, at least in part because of their inability to account for the diversity of chemical species involved in SOA formation. Parameterizations tend to represent SOA constituents as a few lumped products, either with yields and partitioning coefficients derived from chamber studies, or, more successfully, with vapor pressures that reduce progressively in response to parameterized oxidation rates. While empirically based and computationally efficient, neither method contains detailed information about the chemical composition of the SOA. Consequently, aerosol properties cannot be predicted a priori, and it is far from clear that parameterized properties based on chamber experiments are applicable to real atmospheric SOA, for either urban or remote conditions.
ACD scientists Julia Lee-Taylor and Sasha Madronich of the Chemical Processes and Regional Modeling group are using the GECKO-A model (Generator of Explicit Chemistry and Kinetics of Organics in the Atmosphere) to investigate the formation of SOA from an explicit chemistry perspective. The model, developed jointly at NCAR and the University of Paris 12, uses structure-activity relations derived from compilations of laboratory measurements to estimate detailed chemical pathways and kinetics (Aumont et al., 2005) . It predicts the chemical identity of the products as well as their properties relevant to gas/particle partitioning. A typical simulation, starting with 60 precursor hydrocarbons, simulates ~5 million gas-phase reactions among ~1 million species. SOA is produced by applying the partitioning theory developed by Pankow (1994) based on Raoult’s law, in which partitioning depends on the saturation vapor pressure and mole fraction of each contributing compound, and on the total amount of particulate organic mass.
Figure 1 shows results of a simulation from Mexico City, during the 2006 MIRAGE campaign. The model was initialized with observations of about 50 hydrocarbons including alkanes (up to decane), alkenes (up to pentene) and monoaromatics (up to trimethyl benzenes). Larger hydrocarbons (up to 20 carbons) were represented as n-alkanes, based on measurements from Los Angeles (Fraser et al., 1997) scaled to Mexico City decane observations. Diurnal emission profiles were taken from the WRF-Chem model (Tie et al., 2009). The figure includes a 15-hour Eulerian 'spin-up' phase, to 3pm on day 1, after which the air parcel was “released” as Lagrangian outflow diluting with clean background air at a rate of 1/day.
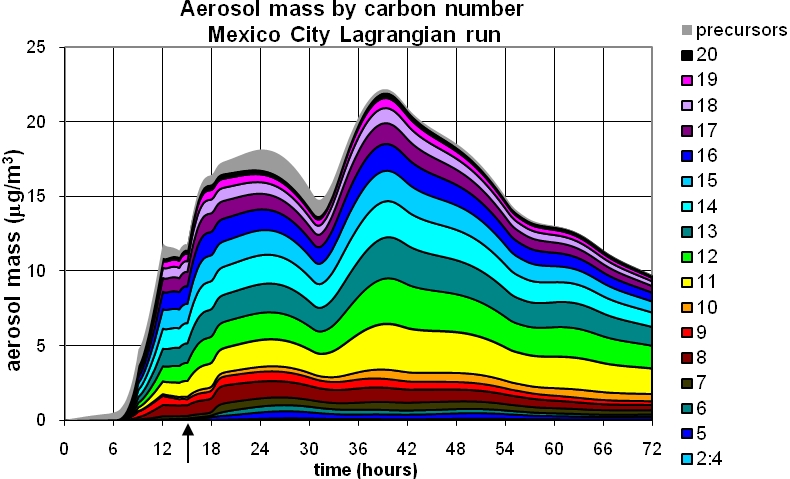
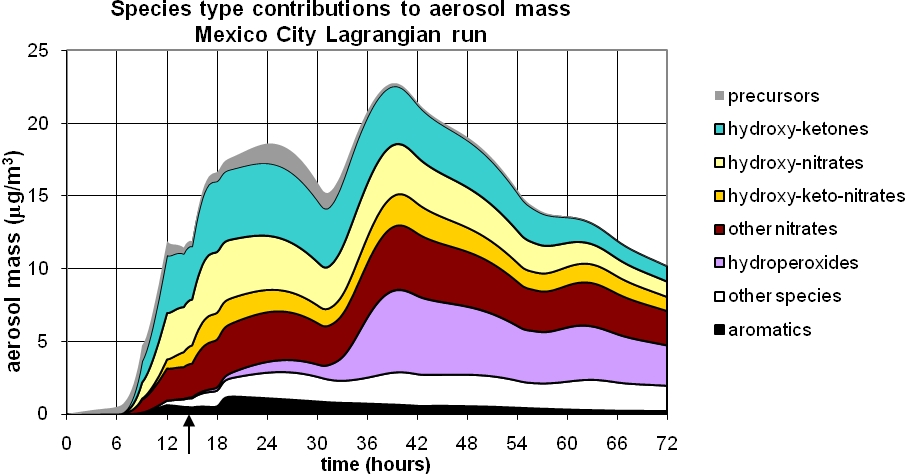
Figure 1. GECKO-A model prediction of organic aerosol in Mexico City during MILAGRO. Top panel: Contributions from partly oxidized organics having carbon chain length indicated in the legend. Bottom panel: Contributions from dominant functional groups. The grey area (top trace of each plot) shows contribution of un-reacted precursor hydrocarbons. Arrow indicates time (3 pm of first day) of parcel release from Eulerian to Lagrangian mode.
Notable points are:
- The calculated SOA concentrations are comparable with observations, and are much larger than predicted with a yield-based 2-product model (e.g. of Volkamer et al., 2006).
- The majority of the SOA molecules are long chains with 11 or more carbons which derive from the long chain alkanes associated with POA (primary organic aerosol) emissions.
- The contribution to SOA from alkanes containing 11-20 carbons is relatively uniform with carbon number, due to the compensating effects, as molecule size increases, of lower concentration and lower vapor pressures.
- Despite the strong dilution, SOA concentrations continue to increase on the second day, and decline relatively slowly on the third day. Thus, evaporation of aging SOA appears to be minimal, and the total mass of SOA produced in the outflow can exceed the mass produced on the first day within the city.
- Major chemical structures in the SOA molecules are hydroxy ketones, hydroxy nitrates, and hydroxy keto-nitrates, most of which arise from the 1,4 H shift isomerization of alkoxy radicals. At later times hydroperoxides become important.
The O/C ratios found in our simulations, 0.2 to 0.3, are somewhat smaller than observed, 0.4-0.9, by DeCarlo et al. (2010), while the simulated N/C ratios are ~0.08, substantially higher than the values of <0.01 reported by Aiken et al. (2008). Our underestimation of the O/C ratio may be due to additional pathways, e.g. from glyoxal (Volkamer et al., 2007) or from the dihydrofurans. It may also indicate that our vapor pressure parameterization is too aggressive, condensing species before sufficient oxidation has taken place. We plan to investigate these pathways in the model.